biom_calc module¶
This module provides methods for calculating various metrics with regards to each OTU in an input OTU abundance table.
arcsine_sqrt_transform¶
Takes the proportion data from relative_abundance() and applies the variance stabilizing arcsine square root transformation:
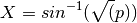
usage: phylotoast.biom_calc.arcsine_sqrt_transform(rel_abd)
-
rel_abd:¶ Refers to a dictionary keyed on SampleIDs, and the values are dictionaries keyed on OTUID’s and their values represent the relative abundance of that OTUID in that SampleID. rel_abd is the output of relative_abundance() function.
-
return:¶ Returns a dictionary keyed on SampleIDs, and the values are dictionaries keyed on OTUID’s and their values represent the transformed relative abundance of that OTUID in that SampleID.
mean_otu_pct_abundance¶
Calculate the mean OTU abundance percentage.
usage: phylotoast.biom_calc.mean_otu_pct_abundance(rel_abd, otuIDs)
-
rel_abd:¶ Refers to a dictionary keyed on SampleIDs, and the values are dictionaries keyed on OTUID’s and their values represent the relative abundance of that OTUID in that SampleID. rel_abd is the output of relative_abundance() function.
-
otuIDs:¶ A list of OTUID’s for which the percentage abundance needs to be measured.
-
return:¶ A dictionary of OTUID and their percent relative abundance as key/value pair.
MRA¶
Calculate the mean relative abundance.
usage: phylotoast.biom_calc.MRA(biomf)
-
biomf:¶ A BIOM file.
-
return:¶ A dictionary keyed on OTUID’s and their mean relative abundance for a given number of sampleIDs.
raw_abundance¶
Calculate the total number of sequences in each OTU or SampleID.
usage: phylotoast.biom_calc.raw_abundance(biomf, sampleIDs=None, sample_abd=True)
-
biomf:¶ A BIOM file.
-
sampleIDs:¶ A list of column id’s from BIOM format OTU table. By default, the list has been set to None.
-
sample_abd:¶ A boolean operator to provide output for OTUID’s or SampleID’s. By default, the output will be provided for SampleID’s.
-
return:¶ Returns a dictionary keyed on either OTUID’s or SampleIDs and their respective abundance as values.
relative_abundance¶
Calculate the relative abundance of each OTUID in a Sample.
usage: phylotoast.biom_calc.relative_abundance(biomf)
-
biomf:¶ A BIOM format.
-
return:¶ Returns a dictionary keyed on SampleIDs, and the values are dictionaries keyed on OTUID’s and their values represent the relative abundance of that OTUID in that SampleID.
transform_raw_abundance¶
Function to transform the total abundance calculation for each sample ID to another format based on user given transformation function.
usage: phylotoast.biom_calc.transform_raw_abundance(biomf, fn=math.log10, sampleIDs=None, sample_abd=True)
-
biomf:¶ A BIOM file.
-
fn:¶ Mathematical function which is used to transform smax to another format. By default, the function has been given as base 10 logarithm.
-
sampleIDs:¶ A list of column id’s from BIOM format OTU table. By default, the list has been set to None.
-
sample_abd:¶ A boolean operator to provide output for OTUID’s or SampleID’s. By default, the output will be provided for SampleID’s.
-
return:¶ Returns a dictionary similar to output of raw_abundance function but with the abundance values modified by the mathematical operation. By default, the operation performed on the abundances is base 10 logarithm.